There are two sub-types of GSD1, we explain the difference between 1a and 1b. Much of the symptoms and management are the same for both.
A general introduction to Glycogen Storage Disease (GSD)
Glycogen Storage Diseases (GSDs) are a group of rare metabolic disorders. To be a fit and healthy individual we must feed our body regularly with a variety of foods and water. This allows growth, provides energy and repairs tissues. The foods that we eat are broken down into smaller packages and either used for growth and repair, stored for when they’re needed for periods between meals or they’re disposed of as waste. Although this explanation describes the basic processes in the body, it is of course much more complex.
Glycogen Storage Diseases affect roughly one child in 20,000. The underlying problem in all GSD’s is the use or storage of glycogen. Glycogen is a complex material that is composed of glucose molecules that are linked together (like a tree with many branches). Most of the cells in the body rely on glucose as their main energy source. The body controls its levels of glucose in the blood very carefully using different hormones. Immediately after a meal, the level of glucose rises and there becomes more sugar in the blood than the body needs at that time. The extra glucose is then moved to the liver and muscles and stored as glycogen, serving as a store for when the body’s sugar level drops below the normal level. We have specific enzymes to help us form or release this energy as it’s needed. Enzymes are protein molecules that speed up chemical reactions in the body.
The moving of glucose to glycogen and back again takes many complicated steps and the body requires enzymes to do this. These enzymes are responsible for creating the glycogen from glucose, moving the glycogen to and from the storage areas within the cells and extracting glucose from the glycogen when needed. A different enzyme is required at each step. People affected with GSD have a problem in one of these many enzymes. The enzyme that’s not working fails to complete its step and the process comes to a stop.
GSD1a – von Gierke disease
GSD1a is also known as Glucose‐6‐phosphatase deficiency. An error in the enzyme called glucose-6-phosphatase (g-6-p) that is mainly found in the liver but also in the kidney and small intestine is the cause of GSD1.
The last step of moving stored glycogen is the change of glucose-6-phosphate into glucose. In GSD1a this step does not happen and so glucose (sugar) cannot be delivered from the liver into the bloodstream.
GSD1b – von Gierke disease
GSD1b is also known as Glucose‐6‐phosphatase translocase deficiency. It is caused by the absence or deficiency of an enzyme called glucose-6‐phosphatase translocase.
For the last step of moving stored glycogen into glucose, glucose-6‐phosphate (g-6-p) must be moved to a certain area of the cell, but because the enzyme that is required for the move is missing or not working properly, the g-6‐p does not arrive in the right place and the last step does not happen and so glucose (sugar) cannot be delivered from the liver into the bloodstream.
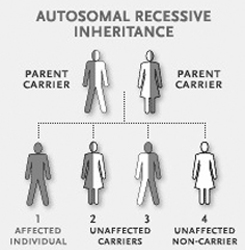
How has my child got this condition?
Glycogen Storage Diseases are genetic. This means that they are not brought about by anything that my have occurred during pregnancy. Genetic disorders are inherited and the pattern in which the child may have developed the condition will now be described.
In GSD1 the fault is inherited from both Mum and Dad and is described as autosomal recessive.
The instructions to make a human being are contained in our genes. For most things we have two copies of the gene; 1 inherited from our mother, and 1 from our father. Each person has 6 or 7 mistakes (mutations) in the thousands of genes, but as we have one working copy of that gene, we are ‘normal’ as it compensates and can do the job of both genes. If when you have a baby you and your partner carry the same genetic mistake there is a 1 in 4 chance with every pregnancy that these two faulty genes will come together, and so the baby has no working copy of the gene. The result is the enzyme is not made correctly as the instructions are faulty, and so the condition develops.